I want to run some sequencing data through the release pipeline that has been analyzed using both the Mutect and Varscan2 variant calling algorithms. Meaning I have two sets of maf files (turned into ssm) on the same donor/sample set, created by the two algorithms. I would like to include the files from both analyses as input to dcc-release, but I’m concerned that doing so would result in some mutations picked up by both algorithms getting counted as two different instances of the same mutation in a single donor.
How have you handled this sort of scenario before? Does the release pipeline account for that sort of input data, or does it expect every instance of a mutation to only appear in the input data once?
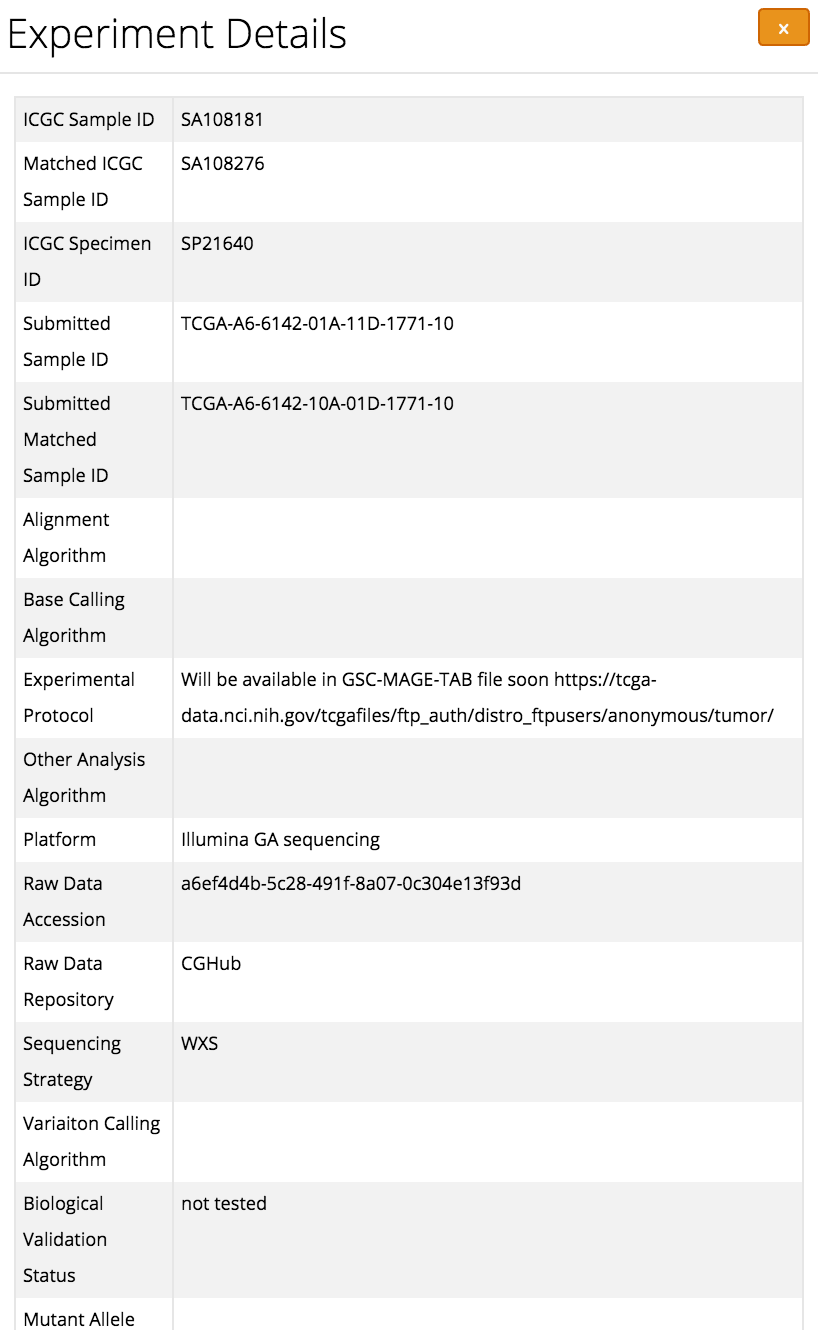
If two variants have the same business key they will be assigned to the same mutation ID. However the end result will show them as two distinct occurrences of that particular mutation with different values for things like calling algorithm and experimental protocol.
Here you can see what the end result for a single occurrence of a mutation would look like in the data portal: